6.11 EMP_multi_analysis
The module EMP_multi_analysis is designed to integrate multiple statistically significant p-values into a single integrated p-value by statistical analysis. Specify the method of joint analysis with the parameters method, including feature, diff_feature_enrich, and same_feature_enrich.
6.11.1 Fusion of statistical results of the same feature from different study cohorts
Different study cohorts are often designed for similar study objective, while some features in these results may be inconsistent, which is especially prevalent in microbial data analysis. To address this, the combining dependent P-values method can integrate multiple statistical results into a single composite result.
🏷️Example:
Microbial data from the two regions were extracted from the MAE object, and difference analysis was performed.
k1 <- MAE |>
EMP_assay_extract('taxonomy') |>
EMP_filter(Region == 'Paris') |>
EMP_diff_analysis(method = 'DESeq2',.formula = ~Group)
k2 <- MAE |>
EMP_assay_extract('taxonomy') |>
EMP_filter(Region == 'Guangzhou') |>
EMP_diff_analysis(method = 'DESeq2',.formula = ~Group)
The p-value fusion method was used to fuse the p-value results of the two differential analyses.
①The parameter
combineMethod provides three methods to fuse P: fisher, edgington and stouffer.②The parameter
action='get' can extract the result of difference analysis.
(k1+k2) |>
EMP_multi_analysis(method = 'feature',combineMethod='edgington',
p.adjust = 'BH')

6.11.2 Fusion of statistical results of the same feature from different study cohorts
🏷️Example:
KO data from the two regions were performed difference analysis and KEGG enrichment.
k1 <- MAE |>
EMP_assay_extract('geno_ko') |>
EMP_filter(Region == 'Paris') |>
EMP_diff_analysis(method = 'DESeq2',.formula = ~Group) |>
EMP_enrich_analysis(pvalue < 0.05,keyType = 'ko')
k2 <- MAE |>
EMP_assay_extract('geno_ko') |>
EMP_filter(Region == 'Guangzhou') |>
EMP_diff_analysis(method = 'DESeq2',.formula = ~Group) |>
EMP_enrich_analysis(pvalue < 0.05,keyType = 'ko')
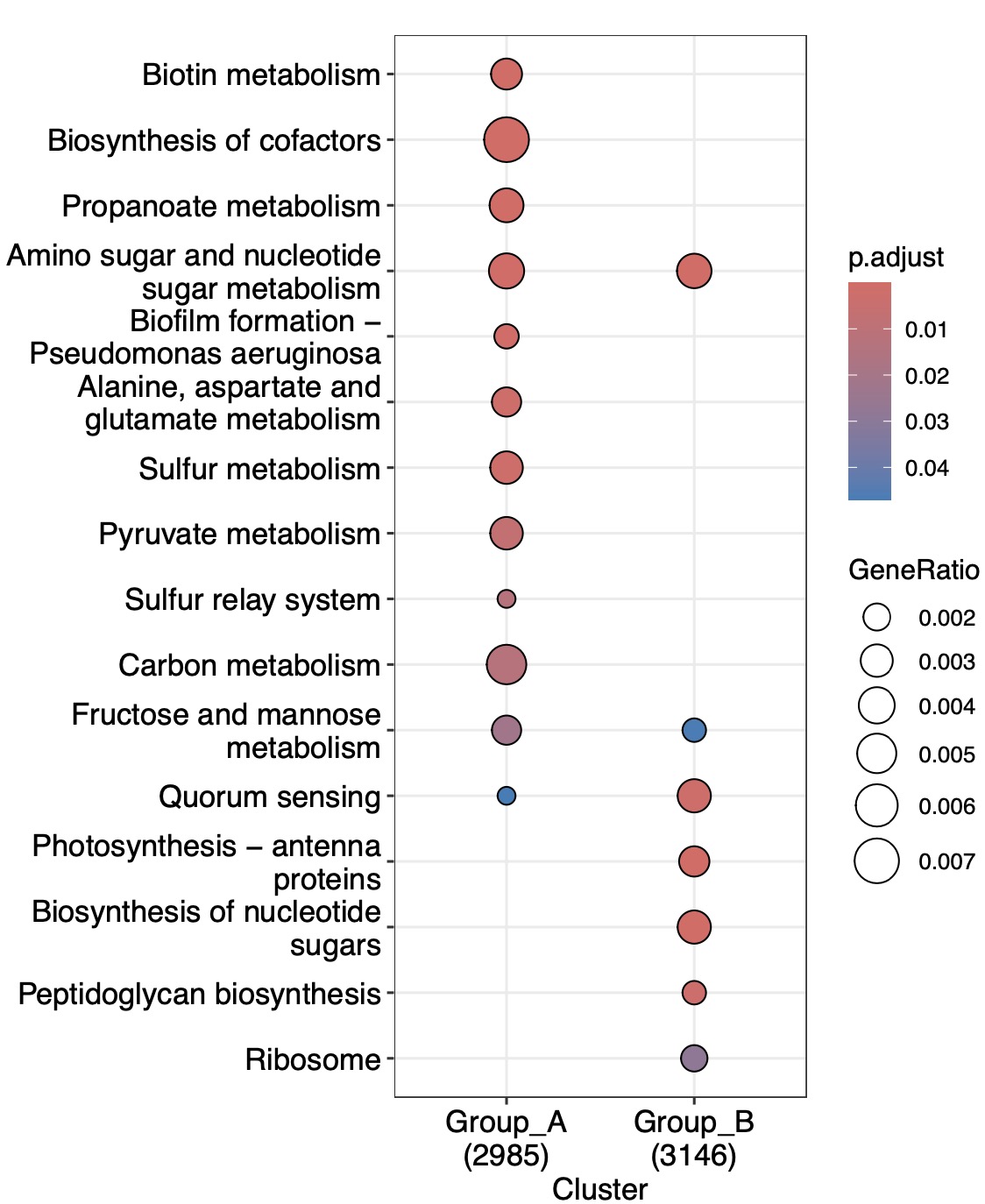
For the enrichment with the same features, this function provides three methods to combine them: enchier, ActivePathways, and mitch。
①When the
enchier method is activated, the combine process is performed by P fusion. By adjusting the parameter combineMethod , it is possible to determine whether the fusion p-value occurs at the feature level or at the enrichment analysis result level.②The
ActivePathways and mitch algorithms rely on their published packages.(k1+k2) |>
EMP_multi_analysis(method = 'same_feature_enrich',
keyType = 'ko',combineFun='ActivePathways')|>
EMP_enrich_dotplot()
6.11.3 Fusion of enrichment results of the different features from the same study cohort
Sequencing analyses of functional genes and metabolomics are occasionally conducted simultaneously within the same cohort, leading to two sets of KEGG enrichment assays that may not always be identical. In such cases, using a Venn diagram alone to identify intersections may not effectively reveal the enrichment relationships between the different omics. To address this issue, this module employs algorithms to integrate KEGG enrichment results across different features in the distinct omics datasets.
🏷️Example:
KO annotation result and metabolomics result were extracted from MAE objects, and KEGG enrichment analysis was completed separately.
k1 <- MAE |>
EMP_assay_extract('geno_ko') |>
EMP_diff_analysis(method = 'DESeq2',.formula = ~Group) |>
EMP_enrich_analysis(pvalue < 0.05,keyType = 'ko')
k2 <- MAE |>
EMP_collapse(experiment = 'untarget_metabol',na_string=c('NA','null','','-'),
estimate_group = 'MS2kegg',method = 'sum',collapse_by = 'row') |>
EMP_diff_analysis(method = 'DESeq2',.formula = ~Group) |>
EMP_enrich_analysis(pvalue < 0.05,keyType = 'cpd')
Use the + sign to combine omics for enrichment analysis.
(k1+k2) |>
EMP_multi_analysis(method = 'diff_feature_enrich') |>
EMP_enrich_dotplot()
